

- 服务与能力
- 生产体系
- 哺乳动物细胞
$我们的专业能力覆盖从哺乳动物细胞培养到生物大分子的发现、开发与 cGMP 生产。依托 6 大新药发现平台、一流的 CMC 开发团队以及完善的供应链体系,我们能够为您的生物药开发提供全流程、一体化的解决方案。
- 哺乳动物细胞表达
$从概念到商业化,为您提供哺乳动物细胞生物药开发的一站式全流程服务。
哺乳动物细胞表达
从概念到商业化,为您提供哺乳动物细胞生物药开发的一站式全流程服务。
- 单克隆抗体
$探索我们针对该类产品提供的全方位药物研发服务
单克隆抗体
探索我们针对该类产品提供的全方位药物研发服务
- 双特异性及多特异性抗体
$探索我们针对该类产品提供的全方位药物研发服务
双特异性及多特异性抗体
探索我们针对该类产品提供的全方位药物研发服务
- Fc融合蛋白
$探索我们针对该类产品提供的全方位药物研发服务
Fc融合蛋白
探索我们针对该类产品提供的全方位药物研发服务
- 抗体片段
$探索我们针对该类产品提供的全方位药物研发服务
抗体片段
探索我们针对该类产品提供的全方位药物研发服务
- 重组蛋白 / 酶 / 细胞因子
$探索我们针对该类产品提供的全方位药物研发服务
重组蛋白 / 酶 / 细胞因子
探索我们针对该类产品提供的全方位药物研发服务
- 抗体偶联药物(ADC)
$探索我们针对该类产品提供的全方位药物研发服务
抗体偶联药物(ADC)
探索我们针对该类产品提供的全方位药物研发服务
- 病毒样颗粒(VLP)
$探索我们针对该类产品提供的全方位药物研发服务
病毒样颗粒(VLP)
探索我们针对该类产品提供的全方位药物研发服务
- 微生物发酵
$全方位 CMC 开发与 cGMP 生产微生物发酵平台。提供基于大肠杆菌及酵母表达系统的质粒 DNA 与重组蛋白生产服务。
- 微生物发酵
$卓越品质,专为微生物发酵来源生物药提供专家级服务。
微生物发酵
卓越品质,专为微生物发酵来源生物药提供专家级服务。
- 抗体片段
$探索我们针对该类产品提供的全方位药物研发服务
抗体片段
探索我们针对该类产品提供的全方位药物研发服务
- 酶
$探索我们针对该类产品提供的全方位药物研发服务
酶
探索我们针对该类产品提供的全方位药物研发服务
- 病毒样颗粒(VLP)
$探索我们针对该类产品提供的全方位药物研发服务
病毒样颗粒(VLP)
探索我们针对该类产品提供的全方位药物研发服务
- 核心能力
- 发现
$涵盖从早期概念至 IND 申报的集成化药物发现平台
- 研究探索
$药明生物提供行业专业知识、最先进的设施和多种抗体生成技术平台,用于发现新型单克隆 双特异性和多特异性抗体、免疫细胞因子和其他生物制剂。
研究探索
药明生物提供行业专业知识、最先进的设施和多种抗体生成技术平台,用于发现新型单克隆 双特异性和多特异性抗体、免疫细胞因子和其他生物制剂。
- 开发
$依托全球规模领先、经验丰富的开发团队,我们拥有卓越的资源、技术与专业实力,致力于以最高效、最具成本效益的方式,驱动您的项目顺利推进至 IND 与 BLA 申报。
- 细胞株工程
$无论是作为独立服务,还是作为我们集成化 CMC 开发平台的一部分,药明生物都能在广泛的生物药领域,为客户提供深厚的专业积淀,以及行业领先的细胞株工程与菌株开发周期。
细胞株工程
无论是作为独立服务,还是作为我们集成化 CMC 开发平台的一部分,药明生物都能在广泛的生物药领域,为客户提供深厚的专业积淀,以及行业领先的细胞株工程与菌株开发周期。
- 分析科学
$我们提供全方位的分析检测服务,在过程控制(IPC)、成品放行及稳定性研究的方法开发领域拥有顶尖的专业实力。此外,我们还支持细胞株构建、工艺及制剂开发、产品表征、可开发性评估,以及其他支持 IND 和 BLA 申报的关键研究。
分析科学
我们提供全方位的分析检测服务,在过程控制(IPC)、成品放行及稳定性研究的方法开发领域拥有顶尖的专业实力。此外,我们还支持细胞株构建、工艺及制剂开发、产品表征、可开发性评估,以及其他支持 IND 和 BLA 申报的关键研究。
- 上下游工艺开发
$我们拥有多个上游与下游工艺开发实验室,支持分批补料、强化分批补料及连续生产工艺的建立与放大。我们的服务涵盖多种生物药类型,贯穿药物研发的早期及后期阶段。
上下游工艺开发
我们拥有多个上游与下游工艺开发实验室,支持分批补料、强化分批补料及连续生产工艺的建立与放大。我们的服务涵盖多种生物药类型,贯穿药物研发的早期及后期阶段。
- 细胞库建库
$我们提供一站式自有细胞库构建与细胞系表征服务,符合全球 GMP 法规及 ICH 指南要求;同时运营超过 20 个 cGMP 级细胞库车间,确保该关键 CMC 开发环节具备充足产能并可按时执行。
细胞库建库
我们提供一站式自有细胞库构建与细胞系表征服务,符合全球 GMP 法规及 ICH 指南要求;同时运营超过 20 个 cGMP 级细胞库车间,确保该关键 CMC 开发环节具备充足产能并可按时执行。
- 生产
$我们在四个国家布局了多座先进且高品质的 cGMP 生产设施,涵盖临床及商业化规模的药物原液(DS)和制剂(DP)生产,能够支持来源于哺乳动物及微生物表达系统的多种生物制品生产。
- 临床原液(DS)GMP生产
$运营多个高质量、先进的临床规模 cGMP 设施,用于生物制药原液(DS)生产,涵盖哺乳动物和微生物两种表达系统。
临床原液(DS)GMP生产
运营多个高质量、先进的临床规模 cGMP 设施,用于生物制药原液(DS)生产,涵盖哺乳动物和微生物两种表达系统。
- 临床制剂(DP)GMP生产
$多个高度灵活的临床规模制剂(DP)生产设施,按照全球监管机构定义的现行药品生产质量管理规范(cGMP)要求,用于生物制剂和注射用制剂的配方、灌装、贴标及包装。
临床制剂(DP)GMP生产
多个高度灵活的临床规模制剂(DP)生产设施,按照全球监管机构定义的现行药品生产质量管理规范(cGMP)要求,用于生物制剂和注射用制剂的配方、灌装、贴标及包装。
- 商业化生产
$药明生物在四个国家拥有多个先进的、高质量的cGMP原液和制剂生产设施。利用多种规格的、经过验证的西林瓶、胶塞和铝盖组合平台,能够在不同的临床阶段和商业化生产规模上进行水针或冻干制剂产品生产。
商业化生产
药明生物在四个国家拥有多个先进的、高质量的cGMP原液和制剂生产设施。利用多种规格的、经过验证的西林瓶、胶塞和铝盖组合平台,能够在不同的临床阶段和商业化生产规模上进行水针或冻干制剂产品生产。
- 检测
$我们在工艺过程检测、产品表征、放行检测及稳定性方法的开发与检测方面具备深厚的专业能力,既可作为一体化生物药开发平台的支持服务提供,也可作为独立项目开展。我们覆盖广泛的分析检测与生物安全检测卓越中心,以及经监管机构批准的质量控制(QC)实验室,是我们为客户提供各项服务的核心支撑。
- 生物药安全检测
$我们拥有符合 EMA、ISO (CNAS) 及 CMA 认证的高质量自有生物安全检测设施,能够为原材料、细胞株及未加工原液提供外源因子筛查,并结合卓越的病毒清除验证能力,为客户提供一站式生物安全检测服务解决方案。
生物药安全检测
我们拥有符合 EMA、ISO (CNAS) 及 CMA 认证的高质量自有生物安全检测设施,能够为原材料、细胞株及未加工原液提供外源因子筛查,并结合卓越的病毒清除验证能力,为客户提供一站式生物安全检测服务解决方案。
- 分析检测
$凭借全方位的检测开发与分析测试能力,我们助力药物研发的全生命周期——从关键表征研究到支持 IND/BLA 申报的各项试验,包括专业的生物分析与法医鉴定。我们致力于为您独特的产品需求提供量身定制的定制化方案。
分析检测
凭借全方位的检测开发与分析测试能力,我们助力药物研发的全生命周期——从关键表征研究到支持 IND/BLA 申报的各项试验,包括专业的生物分析与法医鉴定。我们致力于为您独特的产品需求提供量身定制的定制化方案。
- 卓越中心
$我们的卓越中心(CoE)在产品全生命周期内提供专业的检测支持,旨在加速项目进程,并确保项目在商业化阶段具备完善的分析就绪能力。
卓越中心
我们的卓越中心(CoE)在产品全生命周期内提供专业的检测支持,旨在加速项目进程,并确保项目在商业化阶段具备完善的分析就绪能力。
- 质量标准
- 质量管理
$我们拥有世界一流的质量体系,并在全球各生产基地实现统一标准化管理。我们的质量体系已通过包括美国 FDA、欧洲 EMA、中国 NMPA、日本 PMDA、韩国 MFDS、新加坡 HSA、巴西 ANVISA 及加拿大卫生部(Health Canada)在内的多家全球监管机构认证,支持多种生物药品的生产与检测。
- 全球质量合规部(GQC)
$我们的全球质量与合规团队统筹审计、IT 质量及风险管控,将合规意识贯彻至每一个环节。这确保了我们交付的每一件生物制品都拥有卓越的安全性与疗效,并在执行标准上与您的要求高度对齐。
全球质量合规部(GQC)
我们的全球质量与合规团队统筹审计、IT 质量及风险管控,将合规意识贯彻至每一个环节。这确保了我们交付的每一件生物制品都拥有卓越的安全性与疗效,并在执行标准上与您的要求高度对齐。
- 质量保证
$全球合规体系、全员质量承诺。依托全球生产基地统一的 QA 标准,我们为生物药及疫苗的商业化生产提供稳定、可靠、符合国际主流监管要求的质量支撑。
质量保证
全球合规体系、全员质量承诺。依托全球生产基地统一的 QA 标准,我们为生物药及疫苗的商业化生产提供稳定、可靠、符合国际主流监管要求的质量支撑。
- 质量控制
$我们拥有符合法规要求的自有 QC 实验室,为所有临床及商业化 GMP 生产基地提供全流程支持。我们不仅确保生产前后的产品高质量检测,更对环境监测、清洗验证、仪器生命周期管理、样品/留样管理及审计等关键职能进行全面监督。
质量控制
我们拥有符合法规要求的自有 QC 实验室,为所有临床及商业化 GMP 生产基地提供全流程支持。我们不仅确保生产前后的产品高质量检测,更对环境监测、清洗验证、仪器生命周期管理、样品/留样管理及审计等关键职能进行全面监督。
- 法规事务
$依托深厚的法规专业积淀,我们为客户提供从 CMC 申报资料、药物递交到药物注册的全方位支持。自 2015 年起,我们已成功支持全球客户申报超过550 余项 IND、CTA、BLA、MAA、NDA 及 EUA 申报,完成200 多个 Module 3 CMC 申报件。
法规事务
依托深厚的法规专业积淀,我们为客户提供从 CMC 申报资料、药物递交到药物注册的全方位支持。自 2015 年起,我们已成功支持全球客户申报超过550 余项 IND、CTA、BLA、MAA、NDA 及 EUA 申报,完成200 多个 Module 3 CMC 申报件。
- 技术平台
- 发现
$药明生物提供了先进全面的抗体发现服务,用于创新型抗体的发现、鉴定和筛选提供全方位的服务。
- WuXiBody ® 双特异性抗体平台
$WuXiBody ® 平台是药明生物开发的一个创新的、专有的技术平台,用于扩大双特异性抗体应用范围。基于最新的工程设计,该平台可以加快6-18个月的研发进程,大幅度降低产品的成本。
WuXiBody ® 双特异性抗体平台
WuXiBody ® 平台是药明生物开发的一个创新的、专有的技术平台,用于扩大双特异性抗体应用范围。基于最新的工程设计,该平台可以加快6-18个月的研发进程,大幅度降低产品的成本。
- WuXiHYbrid™ 杂交瘤单克隆抗体研发平台
$WuXiHYbrid™ 是国内领先、世界一流杂交瘤抗体研发平台,突破性提高了抗体新药研发的质量和速度,已为国内外50+客户成功交付超过200个高质量的单克隆抗体研发项目。
WuXiHYbrid™ 杂交瘤单克隆抗体研发平台
WuXiHYbrid™ 是国内领先、世界一流杂交瘤抗体研发平台,突破性提高了抗体新药研发的质量和速度,已为国内外50+客户成功交付超过200个高质量的单克隆抗体研发项目。
- WuXiLiAb™ 噬菌体展示人抗体库
$全人天然抗体库选取60个健康供体, 总数大于6x10e9 的PBMC或CBMC, 每个供体都单独建库并系统QC,保证了文库的高质量和多样性。
WuXiLiAb™ 噬菌体展示人抗体库
全人天然抗体库选取60个健康供体, 总数大于6x10e9 的PBMC或CBMC, 每个供体都单独建库并系统QC,保证了文库的高质量和多样性。
- 开发
$前沿的生物工艺平台与技术,旨在以更快的速度、更高的效率和更具成本效益的方式,推动高质量生物制剂进入临床试验阶段。
- WuXian™ 定制化蛋白生产服务
$依托药明生物行业领先的高通量高表达、纯化和分析技术,提供各类蛋白生产服务,其中包括抗体、双抗、酶和重组蛋白表达。
监管机构资讯
2020Brexit Special Topic: Pharmaceutical Regulatory Schemes of EMA and MHRA Post-BrexitOct. 29, 2020
 Brexit Special Topic: Pharmaceutical Regulatory Schemes of EMA and MHRA Post-Brexit
Brexit Special Topic: Pharmaceutical Regulatory Schemes of EMA and MHRA Post-BrexitWuXi Biologics Regulatory Updates
Quarter 1 2020 – Brexit Special Topic: Pharmaceutical Regulatory Schemes of EMA and MHRA Post-Brexit
Purpose & Disclaimer: This special topic of the newsletter provides a general introduction on Brexit and pharmaceutical regulatory schemes of EMA and MHRA post-Brexit. This is the Regulatory Affairs team’s interpretation of points that may have current or prospective impact to WuXi Biologics. The points listed should neither be considered comprehensive nor exhaustive.
1. General IntroductionBrexit – British exit – refers to the UK leaving the European Union (EU). A public vote (known as referendum) was held in June 2016, when 17.4 million people opted for Brexit, which gave the Leave side 52% of the voting share, compared with 48% for Remain1.
The EU was an economic and political union involving 28 European countries (now 27 after the departure of the UK). Amongst other factors, it allows free trade, as well as free movement of people to live and work in whichever country they choose. The UK joined in 1973 (when it was known as the European Economic Community) and was the first member state to withdraw1.
While the UK has agreed on the terms of its EU departure, years have been taken for both sides to negotiate many aspects of the future UK-EU relationship, including trade and tariffs, law enforcement, aviation standards and safety, supplies of electricity and gas, and licensing and regulation of medicines, etc1. Figure 1 below describes the key milestones for the Brexit process since 20162. On January 31, 2020, the UK formally left the EU. The withdrawal agreement establishes the terms of the UK’s departure from the EU, including a transition period that began on February 1, 2020, and is scheduled to end on December 31, 2020.
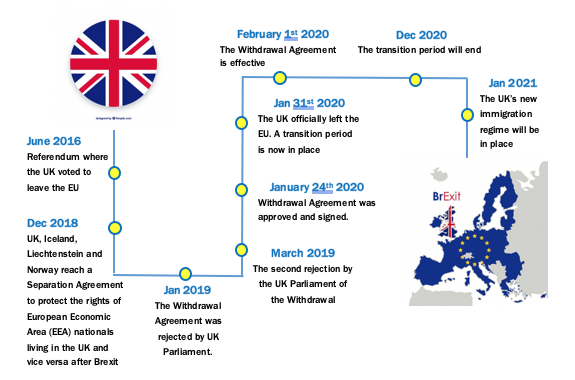
Figure 1 Key Milestones for the Brexit Process Since 20162
2. Pharmaceutical Regulatory Schemes Post-Brexit
2.1 European Medicines Agency (EMA)
The European Medicines Agency (EMA) has made preparations to ensure it continues to deliver on its mission to protect public and animal health throughout the Brexit process.
One of the consequences of Brexit was that the EMA relocated to Amsterdam, the Netherlands, in March 2019. This is in line with Regulation (EU) 2018/1718, which covers EMA’s location and seat3.
In addition, the UK no longer participates in EU institutions after its withdrawal from the EU on January 31, 2020. This means that as of February 1, 2020, no one who represents the UK, or is appointed or nominated by the UK can participate in any EMA scientific committee meeting, working party meeting or in the Agency’s Management Board3.
In preparation for Brexit, the EU27 Member States and EMA redistributed the UK’s portfolio of medicines to other EU Member States. This involved transferring over 370 centrally authorized products to rapporteurs (one of the two members of a committee or working party who leads the evaluation of an application) and co-rapporteurs (one of the two members of a committee or working party leading the assessment of an application) from the EU27 plus Iceland and Norway3.
EMA continues to operate in accordance with the timelines set by its rules and regulations throughout the Brexit process.
2.2 Medicines and Healthcare Products Regulatory Agency (MHRA)
During the transition period (ending in December 2020), EU pharmaceutical law will be applicable to the UK. UK is currently working on its legal basis for pharmaceutical regulations after the transition period.
In addition, under the terms of the Withdrawal Agreement, the UK shall not act as a leading authority for risk assessments, examinations, approvals, and authorizations at the level of the Union or of its’ Member States during the transition period4. This means that the UK can only be a concerned member state (CMS) when participating in the decentralized and mutual recognition licensing procedures during the transition period, while one EU member state will lead the assessment of application as reference member state (RMS)4. UK authorities will remain available to provide expert advice during this period.
3. Regulatory Agencies: Guidance for Sponsors
3.1 Brexit-related Guidance from European Medicines Agency (EMA)
Since 2017, the European Medicines Agency (EMA) and the European Commission (EC) have been providing guidance to help pharmaceutical companies responsible for both human and veterinary medicines prepare for the consequences of Brexit. Below briefly summarizes the EMA’s current recommendations for sponsors of the pharmaceutical industry.
During the transition period, EU pharmaceutical law will be applicable to the UK. This means that pharmaceutical companies have until December 31, 2020 to make the necessary changes to ensure that their centrally and nationally authorized medicines comply with EU law and can remain on the EU market5.
For example, the following entities can still be located in the UK until December 31, 2020:
- marketing authorization holders and applicants;
- orphan designation holders;
- qualified persons for pharmacovigilance (QPPVs);
- veterinary medicines: minor use/minor species (MUMS)/limited markets classification holders;
- companies’ manufacturing and batch release sites.
Each batch of finished product must be certified by a Qualified Person (QP) within the European Economic Area (EEA) before being released for placing on the market in the EEA or for export. Also, the site for batch control needs to be located in the EEA or a country covered by a mutual recognition agreement. Since UK will no longer be treated as part of the EEA after the transition period6, products that only have batch release and quality control testing sites for finished product in the UK will have to change the QP batch release and testing sites to a country located in EEA before December 31, 2020.
3.1.2 Guidance on Centrally Authorized Products (Centralised Procedure)
In the EU, a biological product is centrally authorized by EMA/EC (using Centralized Procedure). EMA and EC are currently updating the Brexit-related guidance documents listed below, and will publish the updates shortly. Sponsors should check EMA’s website about Brexit- related guidance for companies regularly5. For detailed information. Please refer to the two guidance documents listed below:
A. Notice to Stakeholders: Withdrawal of the United Kingdom and EU Rules for Medicinal Products for Human Use and Veterinary Medicinal Products (Last Updated: March 2020)
- This guidance contains information on the legal considerations after the end of the transition period, including issues related to marketing authorizations and related procedures, manufacturing and importation of finished products, Official Control Authority Batch Release (OCABR), orphan medicines, and inspection results amongst other information. This guidance also discusses the provisions for products placed on the market before the end of the transition period.
B. Practical Guidance for Procedures Related to Brexit for Medicinal Products for Human and Veterinary Use within the Framework of the Centralised Procedure (Last Updated: March 2020)
- This document provides procedural and practical guidance regarding submission of changes and related fees. It addresses the implications of the withdrawal agreement and the transition period provided for therein. Key topics include grouping, classification and submission of Brexit-related variations, submission for the transfer of a marketing authorization, notifying the change of Official Medicines Control Laboratory (OMCL) currently in UK, and the impact of the UK’s withdrawal on ongoing applications that include manufacturing sites with GMP certificates issued by UK authorities.
3.1.3 Guidance on Nationally Authorized Products
The Coordination Group for Mutual Recognition and Decentralized Procedures – Human (CMDh) has published information for marketing authorization holders of nationally authorized products. For more information, please refer to: CMDh: Brexit-related information for nationally authorized human medicines.
3.1.4 Submission of Brexit-related Type I Variations
Table 1 below summarizes different types of post-authorization/post-approval changes submitted to the EMA. For reference, types of post-approval submissions to the U.S. FDA are also presented. For detailed information, please refer to classification guidance on minor variations of Type IA, minor variations of Type IB and Major.
Table 1: Different Types of Post-Authorization/Post-Approval Changes for Medicinal Products Submitted to (a) the EMA, (b) the US FDA
(a)
Impact on the Quality, Safety or Efficacy Variation Type Submitted to EMA Minor Type IA Type IAIN Type IB Major Type II (b)
Impact on the Quality, Safety or Efficacy Post-approval Changes Submitted to the U.S. FDA Minor Annual Report (AR) Moderate Changes Being Effected (CBE) in 30 Days/Changes Being Effected Supplements Critical Prior Approval Supplement (PAS) EMA encourages marketing authorization holders to submit Brexit-related type IA and type IB variations as early as possible, to enable EMA to confirm compliance with regulatory and legal requirements by the end of the Brexit transition period5.
According to EU law, the marketing authorization holder, QPPV, pharmacovigilance system master file (PSMF) and certain manufacturing sites need to be based in the EEA for a company to be able to market a medicine in the EU (the UK ceased to be a Contracting Party to the EEA Agreement after its withdrawal from the EU on January 31, 2020 and is therefore no longer part of the EEA). For more information and guidance, please refer to Type-IA Variations: Questions and Answers, and Type-IB Variations: Questions and Answers.
Key questions addressed by these two guidances include:
- When shall I submit my Type-I variation?
- Can I group the submission of Type-I variations? Can they be grouped with other types of variation?
- Is the (co-)rapporteur involved in Type-I variations?
- How shall I present and submit my Type-I variation(s)?
- How will my Type-I variation be handled?
- Can my Type-I be part of work-sharing?
- What should I do in case of an unfavorable outcome for my Type-IA or –IAIN variation?
- How and when will the updated product-information annexes become part of the marketing authorization?
- Who should I contact if I have a question when preparing my application or during the procedure?
Please refer to the links above for detailed answers to these questions.
3.2 Medicines and Health Products Regulatory Agency (MHRA) Guidance on Application for a License to Market a Medicine Post-Brexit
MHRA’s guidance, Apply for a License to Market a Medicine in the UK (last updated: February 2020), discusses how to license a medicine for sale in the UK and Europe post-Brexit, including applications through national, centralized and decentralized procedures4. In summary, the specific application procedure taken depends on the license needed:
- Decentralized procedure is used if the applicant wants to market the medicine in the UK and other named EU countries. The UK can only be a concerned member state (CMS) and cannot act as a rapporteur or co-rapporteur during the transition period, while one other state will lead the assessment of application as the reference member state (RMS). The procedure takes up to 210 days. If the application is approved, the UK and each CMS will issue a national license for the product within 30 days of the approval being granted.
- Mutual recognition procedure is adopted if the applicant already has a national license in 1 or more EU countries but wants to market it in other countries (the UK can only be a CMS during the transition period and cannot act as a rapporteur or co-rapporteur). The application process takes up to 90 days.
- National procedure is used if the applicant wants to market a medicine only in the UK. For first-time applicants, email Data@mhra.gov.uk to obtain a 5-digit company number, and get a PL number from the MHRA Portal before submission. The application process takes up to 210 days.
- Centralized procedure is used when the applicant wants to market certain types of medicines throughout the EU, and a centralized license is mandatory for certain new active substances and biotechnology products. Centralized licenses are granted by the EMA.
4. Reference
- BCC News, Online source, https://www.bbc.com/news/uk-politics-32810887, retrieved on March 13th 2020
- Online source, https://www.fragomen.com/sites/brexit/brexit-key-milestones, retrieved on March 13th 2020
- European Medicines Agency, Brexit: the United Kingdom’s Withdrawal from the European Union, Lasted updated on March 2nd 2020, retrieved from https://www.ema.europa.eu/en/about-us/brexit-united-kingdoms-withdrawal-european-union
- Medicines and Health Products Regulatory Agency, Apply for a Licence to Market a Medicine in the UK, Lasted updated on February 18th 2020, retrieved from https://www.gov.uk/guidance/apply-for-a-licence-to-market-a-medicine-in-the-uk#applying-for-a-licence-during-the-transition-period
- European Medicines Agency, Brexit-related guidance for companies, Lasted updated on December 19th 2019, retrieved from https://www.ema.europa.eu/en/about-us/brexit-uk-withdrawal-eu/brexit-related-guidance-companies
- Frequently asked questions on EFTA, the EEA, EFTA membership and Brexit, online source, retrieved from https://www.efta.int/About-EFTA/Frequently-asked-questions-EFTA-EEA-EFTA-membership-and-Brexit-328676
- WuXiHYbrid™ 杂交瘤单克隆抗体研发平台
- WuXiBody ® 双特异性抗体平台
- 质量保证
- 全球质量合规部(GQC)
- 分析检测
- 临床制剂(DP)GMP生产
- 分析科学
- 开发
- 研究探索
- 抗体片段
- 单克隆抗体
- 哺乳动物细胞表达
- 哺乳动物细胞
- 生产体系
WuXi Biologics
Offering End-to-End Solutions
- 服务与能力
- 生产体系
- 哺乳动物细胞
$我们的专业能力覆盖从哺乳动物细胞培养到生物大分子的发现、开发与 cGMP 生产。依托 6 大新药发现平台、一流的 CMC 开发团队以及完善的供应链体系,我们能够为您的生物药开发提供全流程、一体化的解决方案。

- 返回
- 哺乳动物细胞表达
$从概念到商业化,为您提供哺乳动物细胞生物药开发的一站式全流程服务。
哺乳动物细胞表达
从概念到商业化,为您提供哺乳动物细胞生物药开发的一站式全流程服务。
- 单克隆抗体
$探索我们针对该类产品提供的全方位药物研发服务
单克隆抗体
探索我们针对该类产品提供的全方位药物研发服务
- 双特异性及多特异性抗体
$探索我们针对该类产品提供的全方位药物研发服务
双特异性及多特异性抗体
探索我们针对该类产品提供的全方位药物研发服务
- Fc融合蛋白
$探索我们针对该类产品提供的全方位药物研发服务
Fc融合蛋白
探索我们针对该类产品提供的全方位药物研发服务
- 抗体片段
$探索我们针对该类产品提供的全方位药物研发服务
抗体片段
探索我们针对该类产品提供的全方位药物研发服务
- 重组蛋白 / 酶 / 细胞因子
$探索我们针对该类产品提供的全方位药物研发服务
重组蛋白 / 酶 / 细胞因子
探索我们针对该类产品提供的全方位药物研发服务
- 微生物发酵
$全方位 CMC 开发与 cGMP 生产微生物发酵平台。提供基于大肠杆菌及酵母表达系统的质粒 DNA 与重组蛋白生产服务。
- 返回
- 微生物发酵
$卓越品质,专为微生物发酵来源生物药提供专家级服务。
微生物发酵
卓越品质,专为微生物发酵来源生物药提供专家级服务。
- 抗体片段
$探索我们针对该类产品提供的全方位药物研发服务
抗体片段
探索我们针对该类产品提供的全方位药物研发服务
- 核心能力
- 发现
$涵盖从早期概念至 IND 申报的集成化药物发现平台
- 开发
$依托全球规模领先、经验丰富的开发团队,我们拥有卓越的资源、技术与专业实力,致力于以最高效、最具成本效益的方式,驱动您的项目顺利推进至 IND 与 BLA 申报。
- 返回
- 分析科学
$我们提供全方位的分析检测服务,在过程控制(IPC)、成品放行及稳定性研究的方法开发领域拥有顶尖的专业实力。此外,我们还支持细胞株构建、工艺及制剂开发、产品表征、可开发性评估,以及其他支持 IND 和 BLA 申报的关键研究。
分析科学
我们提供全方位的分析检测服务,在过程控制(IPC)、成品放行及稳定性研究的方法开发领域拥有顶尖的专业实力。此外,我们还支持细胞株构建、工艺及制剂开发、产品表征、可开发性评估,以及其他支持 IND 和 BLA 申报的关键研究。
- 生产
$我们在四个国家布局了多座先进且高品质的 cGMP 生产设施,涵盖临床及商业化规模的药物原液(DS)和制剂(DP)生产,能够支持来源于哺乳动物及微生物表达系统的多种生物制品生产。
- 返回
- 检测
$我们在工艺过程检测、产品表征、放行检测及稳定性方法的开发与检测方面具备深厚的专业能力,既可作为一体化生物药开发平台的支持服务提供,也可作为独立项目开展。我们覆盖广泛的分析检测与生物安全检测卓越中心,以及经监管机构批准的质量控制(QC)实验室,是我们为客户提供各项服务的核心支撑。
- 返回
- 生物药安全检测
$我们拥有符合 EMA、ISO (CNAS) 及 CMA 认证的高质量自有生物安全检测设施,能够为原材料、细胞株及未加工原液提供外源因子筛查,并结合卓越的病毒清除验证能力,为客户提供一站式生物安全检测服务解决方案。
生物药安全检测
我们拥有符合 EMA、ISO (CNAS) 及 CMA 认证的高质量自有生物安全检测设施,能够为原材料、细胞株及未加工原液提供外源因子筛查,并结合卓越的病毒清除验证能力,为客户提供一站式生物安全检测服务解决方案。
- 质量标准
- 质量管理
$我们拥有世界一流的质量体系,并在全球各生产基地实现统一标准化管理。我们的质量体系已通过包括美国 FDA、欧洲 EMA、中国 NMPA、日本 PMDA、韩国 MFDS、新加坡 HSA、巴西 ANVISA 及加拿大卫生部(Health Canada)在内的多家全球监管机构认证,支持多种生物药品的生产与检测。
- 返回
- 全球质量合规部(GQC)
$我们的全球质量与合规团队统筹审计、IT 质量及风险管控,将合规意识贯彻至每一个环节。这确保了我们交付的每一件生物制品都拥有卓越的安全性与疗效,并在执行标准上与您的要求高度对齐。
全球质量合规部(GQC)
我们的全球质量与合规团队统筹审计、IT 质量及风险管控,将合规意识贯彻至每一个环节。这确保了我们交付的每一件生物制品都拥有卓越的安全性与疗效,并在执行标准上与您的要求高度对齐。
- 质量保证
$全球合规体系、全员质量承诺。依托全球生产基地统一的 QA 标准,我们为生物药及疫苗的商业化生产提供稳定、可靠、符合国际主流监管要求的质量支撑。
质量保证
全球合规体系、全员质量承诺。依托全球生产基地统一的 QA 标准,我们为生物药及疫苗的商业化生产提供稳定、可靠、符合国际主流监管要求的质量支撑。
- 技术平台
- 发现
$药明生物提供了先进全面的抗体发现服务,用于创新型抗体的发现、鉴定和筛选提供全方位的服务。
- 返回
- WuXiBody ® 双特异性抗体平台
$WuXiBody ® 平台是药明生物开发的一个创新的、专有的技术平台,用于扩大双特异性抗体应用范围。基于最新的工程设计,该平台可以加快6-18个月的研发进程,大幅度降低产品的成本。
WuXiBody ® 双特异性抗体平台
WuXiBody ® 平台是药明生物开发的一个创新的、专有的技术平台,用于扩大双特异性抗体应用范围。基于最新的工程设计,该平台可以加快6-18个月的研发进程,大幅度降低产品的成本。
- 开发
$前沿的生物工艺平台与技术,旨在以更快的速度、更高的效率和更具成本效益的方式,推动高质量生物制剂进入临床试验阶段。
- 返回
- WuXian™ 定制化蛋白生产服务
$依托药明生物行业领先的高通量高表达、纯化和分析技术,提供各类蛋白生产服务,其中包括抗体、双抗、酶和重组蛋白表达。
WuXian™ 定制化蛋白生产服务
依托药明生物行业领先的高通量高表达、纯化和分析技术,提供各类蛋白生产服务,其中包括抗体、双抗、酶和重组蛋白表达。
- WuXia™ 细胞株构建
$药明生物为多种生物治疗药物提供全面的哺乳动物细胞系开发服务,从客户提供的DNA或蛋白质序列开始,到交付出高产量、高产品质量且稳定的单克隆结束。
- WuXia™ 细胞株构建
- 开发
- 质量保证
- 质量标准
- 生产
- 开发
- 抗体片段
- 单克隆抗体
- 哺乳动物细胞
- 生产体系